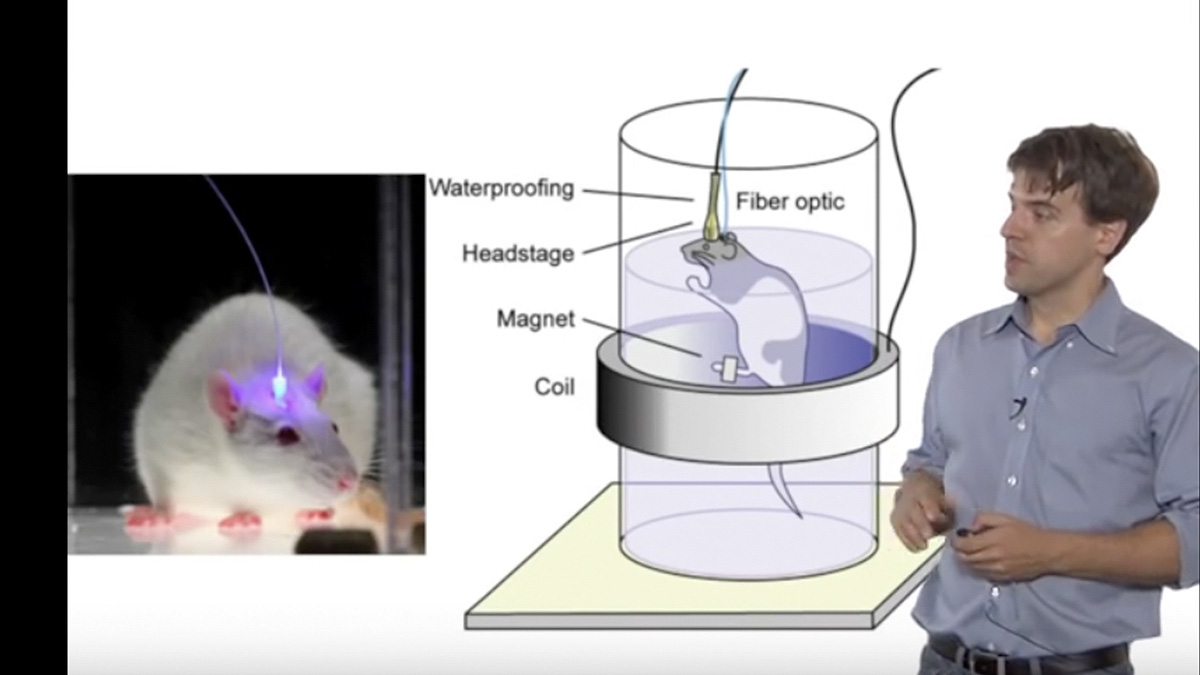


Talk Overview
Mark Schnitzer describes recent work on developing miniature microscopes for deep tissue imaging that can be surgically implemented into living and awake animals. Exciting applications are described for imaging the activity and long term shape changes of single neurons in the brain.
Questions
- What does a GRIN stand for (as in a GRIN lens)?
- Over what time period can a field of neurons in the brain be imaged by this microscope system?
- minutes
- hours
- days
- months
- What is a practical limit of 2-photon imaging?
- 100 nm
- 400 nm
- 1 micron
- 10 microns
Answers
View AnswersSpeaker Bio
Mark Schnitzer

Mark Schnitzer is an Associate Professor in the Departments of Biological Sciences and Applied Physics at Stanford University and an Investigator of the Howard Hughes Medical Institute. His research focuses on understanding learning and memory processes at the level of neural circuits. To this end, his lab has developed techniques capable of observing individual neurons… Continue Reading


Leave a Reply