Talk Overview
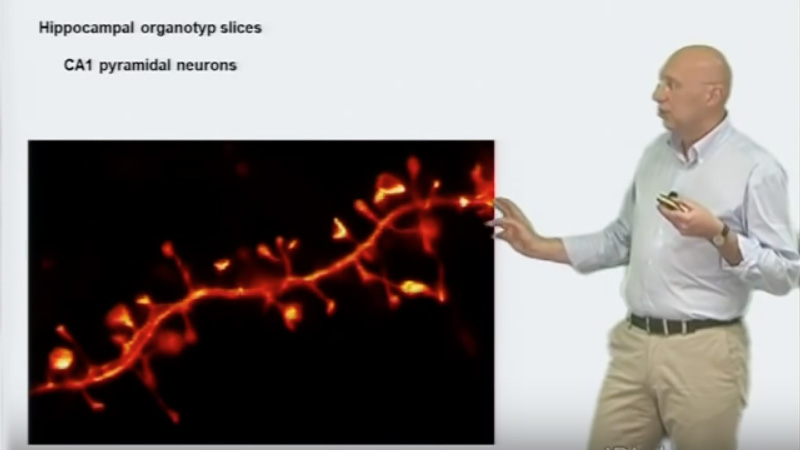
A large family of techniques to achieve super-resolution imaging utilize single molecule switching and localization microscopy. In these techniques, such as STORM, PALM, FPALM, and GSDIM, super-resolution is achieved by first switching all the molecules in the sample to a non-fluorescent state. Individual molecules are then returned to the fluorescent state, imaged, and their position determined to much higher than the diffraction limit. In this talk titled “Super-Resolution: Localization Microscopy,” Bo Huang describes these techniques, dye requirements (photoswitchable fluorescent proteins and small molecule dyes) and how to extend these techniques to 3-dimensional imaging.
Questions
- When localizing a single molecule, what parameter controls the improvement beyond the diffraction limit?
- The number of photons collected
- The pixel size of the camera
- The acquisition frame rate
- The objective magnification
- Localization microscopy relies on what other technique to construct an image?
- Stimulated Emission
- Single Molecule Switching
- Structured illumination
- Confocal Microscopy
- Which of the following influence the final resolution of a single molecule switching image?
- The number of photons collected
- The method used to label the sample
- The number of points recorded
- All of the above
- Which two parameters do you have to trade off when doing live cell single molecule localization microscopy?
- Localization Precision and Speed
- Wavelength and Speed
- Resolution and Speed
- Phototoxicity and Speed
Answers
View AnswersSpeaker Bio
Bo Huang
Dr. Huang’s research focuses on using super-resolution microscopy and single-molecule imaging to understand how proteins form large complexes and how proteins interact to regulate signaling. Huang is an Assistant Professor in Pharmaceutical Chemistry and in Biochemistry and Biophysics at UC San Francisco. Continue Reading

Leave a Reply