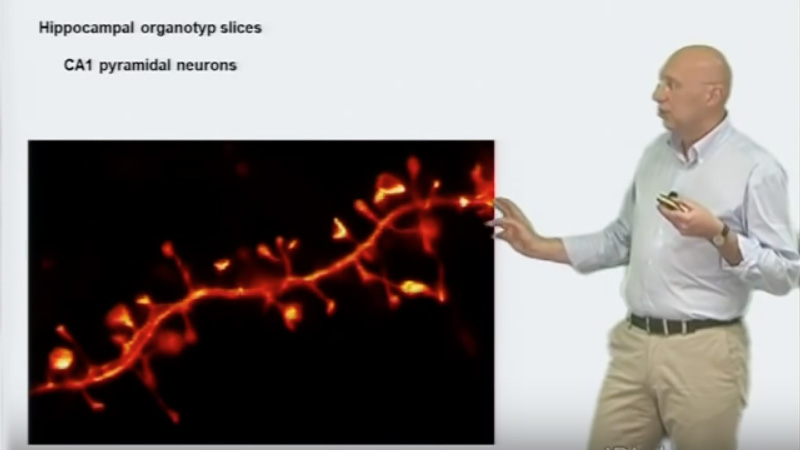
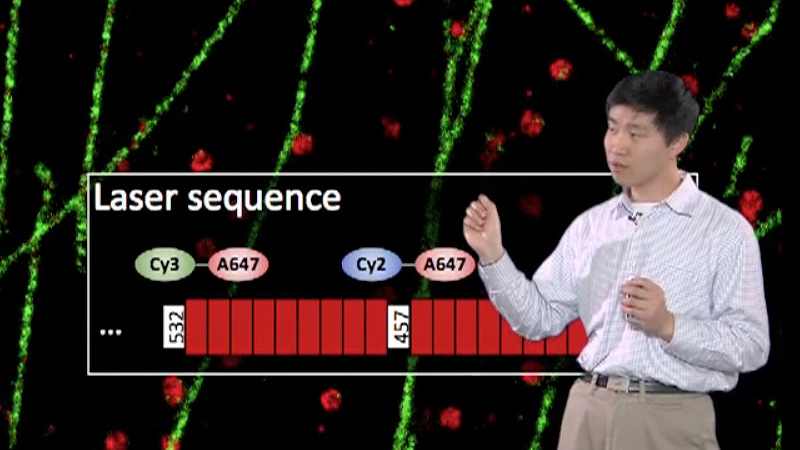
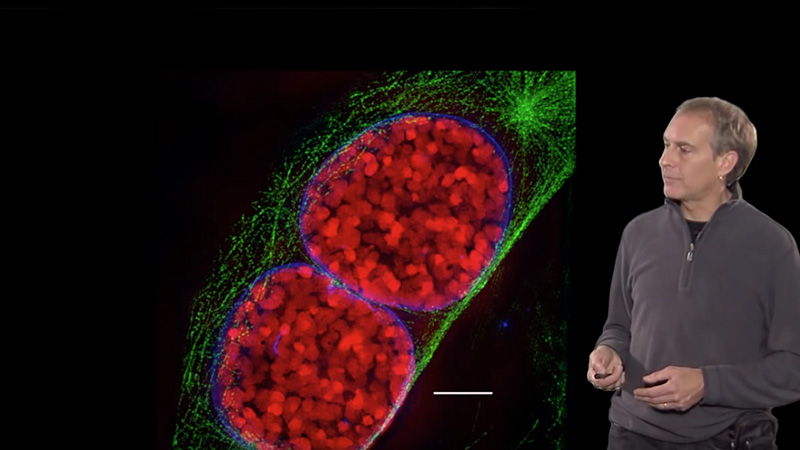
Talk Overview
Total Internal Reflection Fluorescence (TIRF) Microscopy is a technique that only illuminates dye molecules near a surface. In this video, the pioneer of TIRF Microscopy describes what this technique is used for, explains the principles of the evanescent wave, gives many examples of different microscope configurations used in TIRF, and shows how polarized light TIRF can be used to image membrane orientation.
Questions
- What are the main uses of TIRF microscopy?
- Imaging single molecules and cell-substrate contact areas
- Obtaining optical slices throughout living cells
- Imaging cell nuclei
- Imaging developing embryos
- TIRF microscopy reduces background by
- Rejecting fluorescence from out-of-focus areas in the detection path
- Illuminating only the area right near the surface
- Deconvolution
- Only collecting near-field emission
- The evanescent field
- Forms all through the sample
- Can only be explained by quantum mechanical theories
- Decays exponentially and is ⅕ to 1/10th of the wavelength of the excitation light
- Can only be formed by transmitting light through an optical fiber
- TIRF illumination can only be achieved through the objective, True or False.
- TIRF illumination can only be achieved using laser illumination, True or False.
- In comparison to epi-illumination, TIRF
- decreases cell survivability and is unsuited for single molecule spectroscopy
- decreases cell survivability and is well-suited for single molecule spectroscopy
- increases cell survivability and is unsuited for single molecule spectroscopy
- increases cell survivability and is well-suited for single molecule spectroscopy
- What advantages does a high numerical objective have for imaging dye molecules near a surface?
- Allows the capture of more emitted light, including some of the near-field emission
- Has greater magnification
- None
- Captures higher wavelengths better
- DiI embedded in membranes will be excited by polarized TIRF with
- p-polarized light in flat areas and s-polarized light in indentations in the membrane
- p-polarized light in flat areas and p-polarized light in indentations in the membrane
- s-polarized light in flat areas and s-polarized light in indentations in the membrane
- s-polarized light in flat areas and p-polarized light in indentations in the membrane
Answers
View AnswersSpeaker Bio
Dan Axelrod

Dan Axelrod specializes in developing fluorescence microscopy techniques that are useful to cell biologists including total internal reflection fluorescence (TIRF) microscopy, a popular technique for viewing single molecules at surfaces. Axelrod is Professor Emeritus in the Department of Physics and Biophysics at the University of Michigan. Continue Reading

Leave a Reply