

Talk Overview
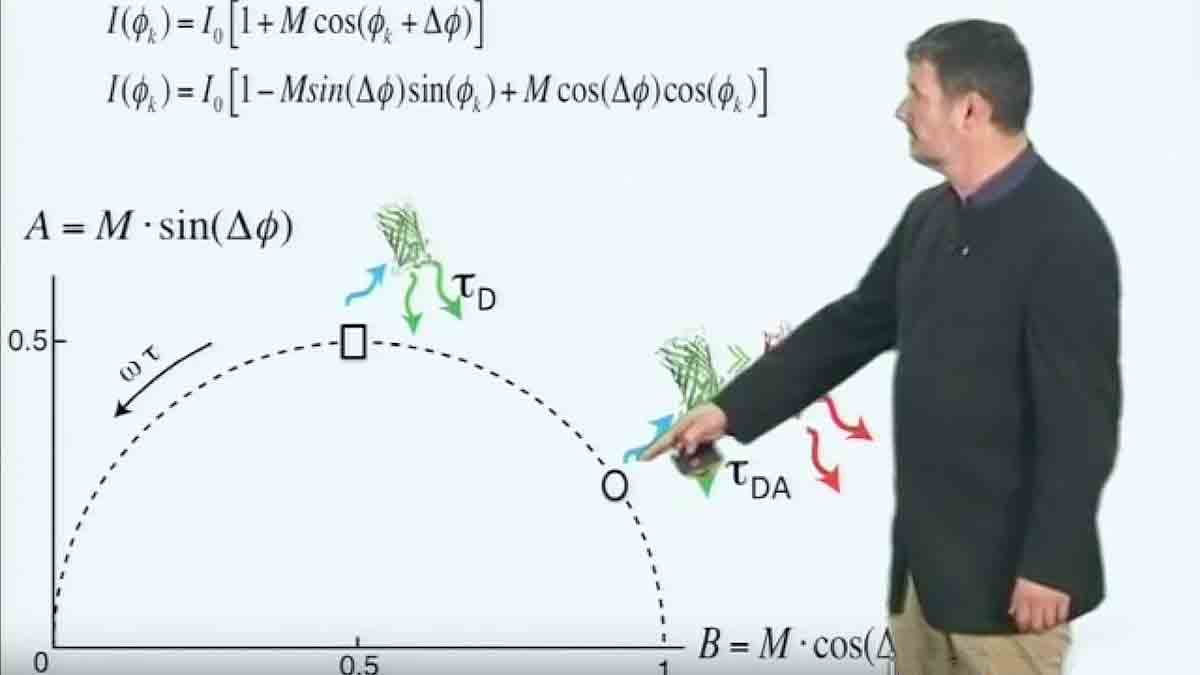
Förster Resonance Energy Transfer (FRET) microscopy is a technique that allows monitoring of interactions between dyes that occur on the nanometer scale. This sensitivity to small changes in distance and orientation make it a popular technique for building biosensors. Here, Philippe Bastiaens describes the physics behind FRET and how FRET can be measured with a microscope.
Questions
- FRET is
- Absorption by acceptor of fluorescence light emitted by the donor
- Nonradiative transfer of energy between nearby molecules with correctly oriented dipoles
- Quenching of fluorescence emitted by acceptor in the presence of donor molecules
- A directly observable physical phenomenon involving fluorescence
- The Förster distance is not a function of
- Orientation factor
- Refractive index
- Overlap between emission of donor and absorption of donor
- Distance between the two molecules
- Förster distances typical for fluorescent proteins are:
- 1-3 Angstrom
- 4-7 Angstrom
- 1-3 nm
- 4-7 nm
- FRET can be measured by ratio imaging when
- Donor and acceptor are part of the same molecule
- Donor and acceptor absorption and emission spectra are known
- There is no overlap in the excitation spectra of donor and acceptor
- There is no overlap in the emission spectra of donor and acceptor
- To estimate sensitized emission, one needs to correct for
- Donor emission at the acceptor emission wavelengths
- Acceptor excitation at the donor excitation wavelengths
- Nothing
- A and B
- FRET measurement by acceptor photobleaching is based on
- Quenching of the acceptor
- Movement of molecules during photobleaching
- Dequenching of the donor
- FRET
- Increases the lifetime of the donor
- Decreases the lifetime of the donor
- Increases the lifetime of the acceptor
- Decreases the lifetime of the acceptor
Answers
View AnswersSpeaker Bio
Philippe Bastiaens

Philippe Bastiaens is Director of the Department of Systemic Cell Biology at the Max Planck Institute of Molecular Physiology and a Professor at the Technische Universitat Dortmund. Bastiaens and his colleagues have developed special fluorescence microscopy techniques such as FLIM and FRET, and use them to explore protein signaling pathways. Continue Reading


Tóth Benedek says
Dear Mr. Philippe,
Why do I have to keep the stochiometry of donor and acceptor constant during ratio imaging?
Sincerely, Tóth Benedek