
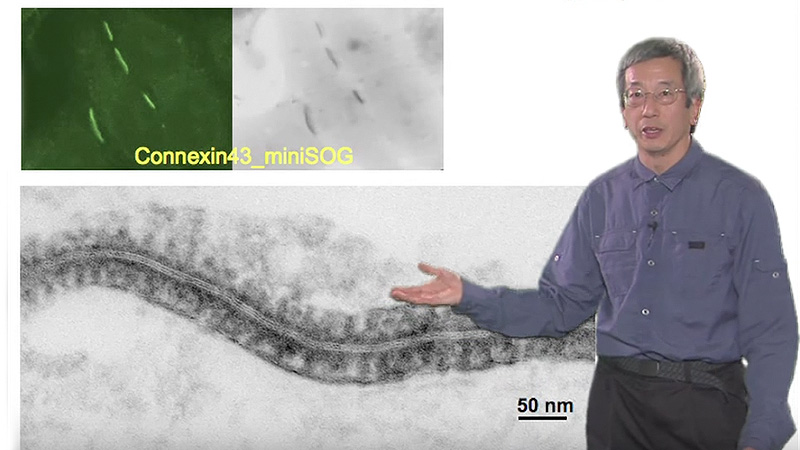

Talk Overview
With all the microscopy techniques now available, which one should you use for your experiment? Here we describe some considerations to take into account when designing a microscopy experiment including: choice of microscope, choice of dye or fluorescent protein, and using confocal, TIRF, or widefield.
Questions
- You would like to obtain a high-resolution image on a thin, 10 µm sample. Which of the following statement(s) is true? (Select all that apply)
- Using a confocal over a widefield microscope will increase the resolution of the image
- Using a high NA over a low NA will increase the image resolution
- Using a spinning disk confocal over a laser-scanning confocal will be preferable if this is a live sample
- None of the above
- In his talk, Dr. Thorn explains that the resolution of an image can be calculated based on the following mathematical formula: dmin = 0.61 λ/ N.A. Would you agree or disagree with the following statement: “The higher the NA, the higher the image resolution.” Explain why.
- You are in charge of the microscopy facility at your institution. An investigator comes to you and explains that, after several months of research, she finally identified a lab which has a transgenic mouse colony expressing mCherry fusion proteins for the gene she is researching. mCherry is a monomeric red fluorescent protein (RFP) that is excited with a 561 nm yellow-green laser. However, you do not have this laser line at your facility. Should you recommend that this investigator not move forward with her study until she finds transgenic mice that match the laser lines available at your facility? Explain your answer.
- You are in the process of designing an experiment which will involve long-term, high-resolution 3D live cell imaging on a 10 µm sample, and involving fusion proteins with mCherry. List 3 pieces of equipment that you will need to check for before you begin your study.
- In the list below, choose the steps that could improve data analysis in the long run:
- Using fewer than 4 dyes at onc
- Determining whether protein localization is impacted by fixing, mounting or labeling prior to beginning your study
- Reducing the amount of light to which you are exposing your samples
- Including controls that will correct for variations during data collection
- None of the above
Answers
View AnswersSpeaker Bio
Kurt Thorn

Kurt Thorn is an Assistant Professor of Biochemistry and Biophysics at UCSF and Director of the Nikon Imaging Center – a facility that provides cutting edge light microscopy equipment to UCSF researchers. Kurt can be followed on his blog at http://nic.ucsf.edu/blog/. Continue Reading


Leave a Reply